




Chiba Medical J. 101E:33-38, 2025
doi:10.20776/S03035476-101E-2-P33
〔 Chiba Medical Society Young Investigator Award Minireview 〕
Maya Kitajima1,2), Kazuko Kita2), and Atsushi Kaneda2)
1) School of Medicine, Chiba University, Chiba 260-8670.
2) Department of Molecular Oncology, Graduate School of Medicine, Chiba University, Chiba 260-8670.
(Received February 27, 2025, Accepted March 10, 2025, Published June 10, 2025.)
p53 plays an important role as one of the most well-known tumor suppressors. Under various stresses, p53 is stabilized and activated, and exerts functions involved in tumor suppression, including the induction of cell cycle arrest, apoptosis, and senescence. Among these, the induction of apoptosis is very important for preventing tumorigenesis. Stabilization of p53 is regulated by the ubiquitin ligase MDM2 or other proteins such as pARF14. In various cancer cells, TP53 gene mutations are found, and the loss of tumor-suppressive functions and gain of oncogenic functions in mutant p53 have been reported to lead to genomic instability and cancer progression. Herein, we summarize the mechanisms of apoptosis induction and the roles of p53 in this process, in which p53 has transactivation-dependent or transactivation-independent functions. In addition, we summarize the regulators of p53 and the mechanisms of p53 stabilization and activation, cancer-associated TP53 mutations, and functions of mutant p53. This review contributes to the understanding of the tumor-suppressive function of p53 and the significance of disrupted functions of mutant p53 as a target for therapeutic strategies.
p53; tumor suppressor, transcription factor, apoptosis, mutant p53
p53, one of the most well-known tumor suppressors, was discovered in 1979, and its functions were clarified between 1989 and 1990[1,2]. Induction of cell cycle arrest, apoptosis, and senescence is the earliest discovered functions of p53. A wide variety of stress signals, such as DNA damage, oxidative stress, and oncogene activation, can induce p53 to perform these functions. Notably, upon DNA damage, p53 is stabilized and activated to arrest the cell cycle and repair the damaged DNA. If the damage is too severe for repair, apoptosis and senescence are induced by p53 to eliminate impaired cells. The loss of p53 function, including inactivating mutations of p53, leads to a failure to eliminate impaired cells, genomic instability, and cancer development[3]. Approximately half of human cancers have inactivating mutations in p53, and most of the remaining have wild-type p53 without mutations, but with the function inhibited by increasing its inhibitors, reducing its activators, or inactivating its downstream targets, making p53 a central player in cancer research[4].
p53 is an essential tumor suppressor that replies to various stress signals by coordinating cellular responses, including cell cycle arrest, senescence, and apoptosis, all of which are involved in tumor suppression. Recent studies have revealed the role of p53 in adjusting other cellular processes that affect tumor suppression, including the regulation of metabolism, autophagy, and the oxidative condition of the cell, along with the suppression of stem cell maintenance, invasion, metastasis, and communication within the tumor microenvironment[5,6]. Among these, the induction of apoptosis is very important for preventing tumorigenesis. Apoptosis is an important mechanism for maintaining cellular homeostasis and preventing the onset of diseases, such as cancer. When the apoptosis induction mechanism is distracted by mutations in p53, the risk of cancer development increases significantly, and the restoration of p53 function has become a target for cancer therapy[7-9]. This review summarizes the role of p53 in apoptosis, regulation of p53 activity, and carcinogenesis when p53 function is distracted.
p53 is best described as a transcription factor that binds to specific DNA sequences and transactivates several genes with various functions including apoptosis[4]. By binding to specific DNA sequences and regulating the transcription of apoptosis-related genes, p53 is involved in both the intrinsic and extrinsic apoptosis-inducing pathways. Although the intrinsic pathway is initiated in response to distinct cellular stressors such as DNA damage and endoplasmic reticulum stress, the extrinsic pathway is triggered by external signals from other cells[10].
p53 induces apoptosis via the intrinsic pathway, mostly by direct transcriptional activation of pro-apoptotic BH3-only proteins of the BCL-2 protein family, such as PUMA and NOXA, which is also called the BCL-2-regulated or mitochondrial apoptotic pathway (Fig. 1)[11]. BH3-only proteins bind to anti-apoptotic BCL-2 proteins (BCL-2, BCL-XL, MCL-1, BCL-W, and BFL1) and inhibit their activity, leading to the activation of BAX and BAK, other pro-apoptotic proteins in the BCL-2 protein family[12]. Activation of BAX/BAK increases mitochondrial permeability and causes the release of cytochrome c. Cytochrome c binds to apoptotic protease-activating factor 1 (APAF1) and pro-caspase-9 to form a complex called an apoptosome, followed by the activation of caspase-9 and effector caspases, including caspase-3 and -7[10]. Activated caspase-3 cleaves the inhibitor of caspase-activated DNase (ICAD), releasing and activating CAD, and the activated CAD executes DNA fragmentation. At the same time, activated caspase-3 cleaves BCL-2 and BCL-XL, causing them to lose their anti-apoptotic functions[13]. Caspase-7 also has similar functions in apoptosis, but with some distinctions from those of caspase-3[14]. Apoptotic cells with fragmented DNA are phagocytosed and eliminated by nearby cells. p53 also transactivates the genes involved in the intrinsic apoptotic pathway, including BAX and APAF1[11] (Fig. 1).
p53 also has biological activities in the intrinsic apoptotic pathway that are transcription independent (Fig. 1). In this pathway, p53 translocates to the mitochondria and promotes mitochondrial permeabilization by interacting with the anti- and pro-apoptotic BCL-2 family proteins and mediates the release of cytochrome c. Thus, p53 is involved in the activation of the intrinsic apoptotic pathway, and its function is also an important part of p53-induced apoptosis[4,15,16].
p53 is involved not only in the intrinsic pathway but also in the extrinsic pathway, which is also called the death receptor pathway (Fig. 1). In the extrinsic pathway, death receptors, including FAS and TNF-related apoptosis-inducing ligand receptor 2 (TRAIL-R2/DR5)[4], are clustered according to their cognate ligands[10]. FAS and TRAIL-R2 /DR5 are p53 target genes[11]. The cluster formation of death receptors and ligands leads to the recruitment and stabilization of a conformer of the adapter proteins FAS-associated death domain (FADD) and TNF receptor-associated death domain (TRADD), which recruits and activates caspase-8[10]. Activated caspase-8 promotes apoptosis via two parallel pathways. The first is through the direct cleavage and activation of caspase-3 and caspase-7, and the second is through the cleavage of BID (Fig. 1), an apoptosis-promoting BCL-2 family protein. Cleaved BID translocates to the mitochondria and induces cytochrome c release, which in turn activates caspase-9 and caspase-3. Thus, the exogenous pathway is not completely independent of the endogenous pathway but is interrelated and converges on a common execution phase[10].
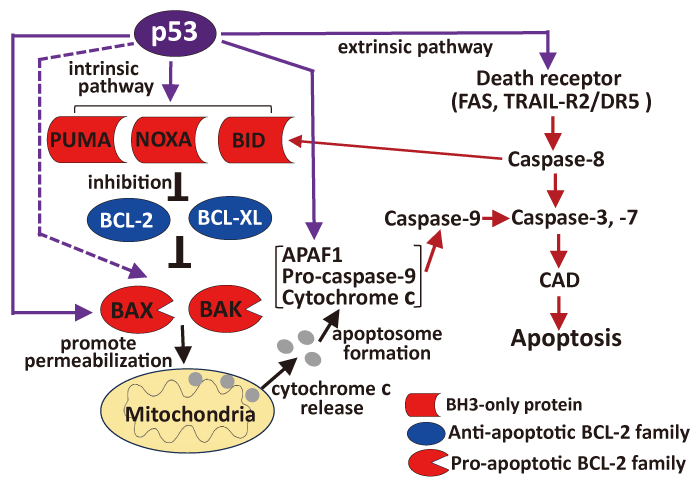
Fig. 1 Overview of p53-related apoptosis. Model depicting intrinsic and extrinsic apoptotic pathways regulated by activated p53 Purple arrows indicate transactivation-dependent upregulation by p53, broken purple arrow indicates transactivation-independent activation by p53, and brown arrows indicate activation of target protein.
In unstressed non-transformed cells, p53 protein level is very low and often undetectable because p53 can be targeted by the E3 ubiquitin ligase MDM2 for proteasomal degradation[11]. In addition, MDM2 can bind to transcriptional activation domain of p53 and inhibit its ability for target gene regulation and antiproliferative effect[17]. However, in response to various stresses, p53 is released from the negative regulator MDM2, thereby stabilizing and activating p53. For example, when cells are exposed to DNA-damaging stress e.g. DNA double-strand breaks or stalled DNA replication forks, ataxia telangiectasia mutated (ATM) kinase or ataxia telangiectasia and RAD3-related (ATR) kinase is activated, respectively. Then p53 is phosphorylated at serine15 by the kinases, displaced from MDM2, and stabilized[5]. Phosphorylation of p53 can occur at several sites and the phosphorylation events are critical for p53 stabilization and activation. Once stabilized, p53 accumulates in the nucleus and is activated as a transcription factor. As a feedback regulator of stress response, p53 induces MDM2 expression[4].
MDM2 is regulated by phosphorylation, auto ubiquitination[18], or binding to p14ARF, which inhibits MDM2 activity and indirectly hinders p53 degradation [19,20]. The p14ARF-MDM2-p53 pathway is disrupted in cancer development. In addition, the MDM2 family member MDMX, also called MDM4, associates with MDM2 via the C-terminal RING finger domain and enhances the E3 ubiquitin ligase activity of MDM2, thereby promoting the degradation of p53[21]. MDMX lacks E3-ligase activity but is still crucial for regulating p53 transcriptional activity, both by binding to and suppressing p53, as well as by regulating MDM2[6]. Herpes virus-associated ubiquitin specific protease (HAUSP) modulates the p53-MDM2 network dynamics in response against DNA damage by de-ubiquitinating both p53 and MDM2[22]. Moreover, there are E3 ubiquitin ligases regulating p53, such as COP1, Pirh2, Arf-Bp1, Topors, CARPs37, and Synoviolin, other than MDM2, though the role of these E3 ligases in regulation of levels of p53 needs further confirmation, e.g. in vivo genetic experiments[23]. These findings suggest that MDM2-independent pathways are involved in regulating p53 activity.
Most mutations in the TP53 gene found in cancer cells are point mutations in the DNA-binding domain (DBD), highlighting the importance of DNA binding for the tumor-suppressive function of p53[11]. The central core spanning residues 100–300 of p53 contains the DBD (Fig. 2) responsible for the sequence-specific binding of p53 to response elements in DNA (p53 DNA REs)[24]. Most cancer-associated TP53 mutations are missense mutations within this domain, disrupting DNA binding and impairing the tumor-suppressive function. Although other mutations, such as truncating, inframe, and splice mutations, have also been reported, missense mutations in the DBD account for approximately 80% of all TP53 mutations (Table 1). These missense mutations can be classified into two types: DNA contact mutants, which alter residues crucial for direct contact with p53 DNA REs, and structural mutants, which affect protein folding. The six most common mutation hotspots in cancer are R175, G245, R248, R249, R273, and R282 (Table 1). Furthermore, by forming complexes with wild-type p53, mutant p53 proteins show dominant-negative effects that could play important roles early during transformation even though wild-type TP53 allele is retained in cells[5,11,25].
Beyond disrupting DNA binding, some of these mutations confer oncogenic properties to p53, potentially contributing to the promotion of tumor cell invasion, migration, proliferation, and metastasis [6,24,26,27]. These findings suggest that mutant p53 may elicit not only loss-of-function on tumor suppression but also gain-of-function on oncogenicity that promote cancer development[28]. One key aspect of this gain-of-function is that mutant p53 interferes with the intramolecular autoactivation of MDM2 and inhibits its E3 ligase activity of MDM2, resulting in accumulation of mutant p53 and contributing to tumorigenesis[29,30]. Additionally, mutant p53-expressing mice exhibit tumor metastasis at an increased frequency compared with those without p53, showing further evidence of its gain-of-function role in cancer progression[29].

Fig. 2 p53 protein domain structure.
Table 1 Different mutation types and their effects on tumor development

p53 induces apoptosis through both the intrinsic and extrinsic pathways and is critical for preventing tumorigenesis. p53 acts in both transcription-independent and -dependent manner. Mutant p53, which leads to loss-of-function, dominant-negative effects, and gain-of-function, is an essential target for cancer therapeutic strategies. Therefore, further studies on mutant p53 are required.
M. K. wrote the original draft of the manuscript. K. K. reviewed and edited the manuscript. A. K. conceptualized, reviewed, and edited the manuscript. All the authors have read the final version of this manuscript.
A. K. is a member of the Editorial Board of the Chiba Medical Journal. The authors declare that there are no other conflicts of interest.
Not applicable.
Not applicable.
The authors thank all laboratory members and collaborators who supported this study.
Address correspondence to Dr. Atsushi Kaneda.
Department of Molecular Oncology, Graduate School of Medicine,
Chiba University, 1-8-1, Inohana, Chuou-ku, Chiba 260-8670, Japan.
Phone/Fax: +81-43-226-2039.
E-mail: kaneda@chiba-u.jp
