




Chiba Medical J. 101E:27-32, 2025
doi:10.20776/S03035476-101E-2-P27
〔 Chiba Medical Society Award Review 〕
Akiyuki Uzawa
Department of Neurology, Graduate School of Medicine, Chiba University, Chiba 260-8670.
(Received December 23, 2024, Accepted January 21, 2025, Published June 10, 2025.)
Myasthenia gravis (MG) is an inflammatory disorder of the neuromuscular junction, which is primarily caused by autoantibodies against the nicotinic acetylcholine receptor (AChR). Achieving remission in MG with standard care is difficult and there is a risk of adverse events because long-term immunosuppression is usually required. Thus, the development of more effective and safer treatments is needed. Based on the concept of“ selective reductions of pathogenic antibodies and pathogenic immune cells without suppressing normal immunity,” we developed AChR-Fc, a fusion protein of the immunoglobulin G1 Fc region and the AChR alpha 1 subunit extracellular domain. AChR-Fc is believed to exhibit two mechanisms of action: selective neutralizing activity against AChR antibodies and cytotoxic activity against AChR antibody-producing B cells. This AChR-Fc fusion protein represents an innovative treatment that may be effective against MG, although future clinical trials will be required.
myasthenia gravis, fusion protein, novel treatment, B cell, animal model
Myasthenia gravis (MG) is an autoimmune disease of unknown etiology, in which autoantibodies are produced against proteins on the neuromuscular junction[1]. Autoantibodies against acetylcholine receptors (AChR) are detected in approximately 80% of MG cases[1]and against muscle-specific tyrosine kinase in approximately 5% of MG cases. AChR antibodies are thought to interfere with neuromuscular transmission by inhibiting the binding of acetylcholine to AChR, inducing the internalization of AChR, and/or destruction of AChR via complement activation[2]. In particular, complement-mediated destruction of the motor endplate is considered a primary factor in the pathogenesis of MG[3,4]. MG is characterized by muscle weakness accompanied by fatigue and fluctuation[1]. The symptoms include ptosis, diplopia, dysarthria, dysphagia, and muscle weakness in the limbs, neck, and respiratory muscles. Non-motor symptoms, including alopecia areata, taste disorders, pure red cell aplasia, neuromyotonia, myocarditis, and immunodeficiency, are rarely confirmed, particularly in thymoma-associated MG[5,6]. A nationwide Japanese epidemiological survey in 2017 revealed that the number of MG patients in Japan was 29,210 (95%CI: 26,030–32,390) with a prevalence of 23.1 per 100,000 individuals (95%CI: 20.5–25.6)[7].
Currently, administration of steroids and/or immunosuppressants and thymectomy are standard treatment for patients with AChR antibody-positive MG; however, it is difficult to maintain a remission status[8,9]. In addition, there are concerns about side effects and worsening quality of life associated with long-term use of steroids[9]. Plasmapheresis and high-dose intravenous immunoglobulin administration may be performed as rescue therapy to remove or neutralize AChR antibodies[10]. In addition, early fast-acting treatment, which is defined as a treatment with plasmapheresis, intravenous immunoglobulin and/or intravenous high-dose methylprednisolone during the early phases of treatment, can provide long-term benefits (e.g. good prognosis) for MG patients[11]. In recent years, new biological drugs have been approved in Japan, such as complement inhibitors (eculizumab, ravulizumab, and zilucoplan) and neonatal Fc receptors inhibitors (efgartigimod and rozanolixizumab)[12]. Complement inhibitors are primarily utilized for patients with AChR-antibody-positive MG, particularly in refractory cases, which do not respond well to immunotherapies. FcRn inhibitors work by reducing blood IgG levels, including pathogenic autoantibodies, making them a promising new therapeutic strategy for IgG autoantibody-mediated autoimmune diseases, such as MG, in which pathogenic IgG antibodies play a central role in pathogenesis. Treatments with FcRn inhibitors are often referred to as “chemical plasma exchange” because of their mechanism of action. These biological therapies are very expensive; thus, refractory cases are generally candidates for treatment. Although the number of treatment options for MG, including early fast-acting treatment and biological treatments, has gradually increased, some patients are not able to achieve their treatment goals[8,13]. It has been reported that 21% of patients were refractory in a Japanese registry study[8]. The therapeutic effect of the available treatment remains insufficient and there are concerns about side effects associated with immunosuppressing treatments. Therefore, the development of new therapies, which are more effective and safer than the current treatments is necessary.
We developed a fusion protein, AChR-Fc, as a novel candidate and innovative therapy for MG[14]. In this review, we discuss the characteristics and effects of AChR-Fc as well as its prospects.
AChR-Fc is a fusion protein created by transfecting a recombinant plasmid containing the human AChR α1 subunit extracellular domain and the Fc region of human IgG1 joined by a linker amino acid sequence into Chinese hamster ovary cells[14]. The fusion protein AChR-Fc is believed to act through two mechanisms: a neutralizing effect of AChR antibodies by acting as a decoy via its AChR structure, and a cytotoxicity effect by the effector cells against AChR antibody-producing pathogenic B cells through its Fc region (Fig. 1). AChR-Fc is innovative because it theoretically selectively suppresses AChR antibodies and pathogenic B cells, which play the central role for MG immunopathogenesis, without affecting normal immune function. These characteristic mechanisms render it more useful compared with conventional therapies in terms of efficacy and safety. We examined the therapeutic effects of this fusion protein on MG in vitro and in vivo[14].
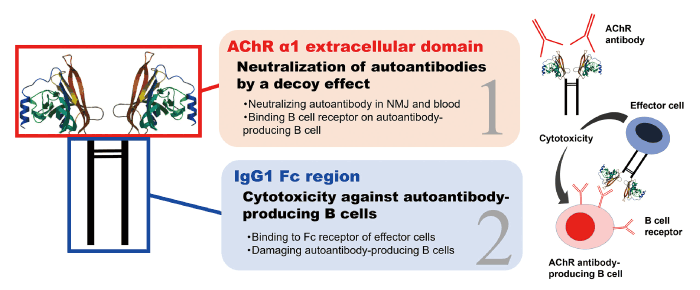
Fig. 1 Structure and mechanism of action of the AChR-Fc fusion protein. AChR-Fc is a fusion protein that contains the human AChR (acetylcholine receptor) structure and the Fc region of human IgG1. It is thought to have two mechanisms: neutralizing activity of AChR antibodies through the AChR structure and antibody-mediated cytotoxicity against AChR antibody-producing pathogenic B cells via the Fc region. NMJ = neuromuscular junction
A binding assay was performed between AChR-Fc and mab35 which is a hybridoma (TIB-175) -derived rat AChR monoclonal antibody. AChR-Fc was fixed on a sensor chip, mAb35 was added, and a dose-dependent binding of mab35 was observed[14]. Next, a binding assay was performed using AChR-Fc and AChR antibodies from MG patients. IgG fractions purified from patients with AChR antibody-positive MG were incubated with AChR-Fc and dose-dependent binding was confirmed[14]. Conversely, binding was not observed using IgG from normal control subjects. The results suggest that antibodies against the α1 subunit of AChR are present in patients with AChR antibody-positive MG and demonstrate that the AChR-Fc fusion protein binds to AChR antibodies in a concentration-dependent manner and exhibits autoantibody neutralizing activity as a decoy in vitro[14].
The binding and cytotoxic effect of AChR-Fc against pathogenic autoantibody-producing hybridomas were examined. TIB-175, which is a rat AChR α1 subunit antibody-producing hybridoma, expresses an AChR antibody as a B cell receptor on its surface. The binding of TIB-175 to AChR-Fc was confirmed by flow cytometry[14]. Because binding was not observed with other hybridomas, AChR-Fc was considered to bind specifically to AChR antibody-producing cells via the B cell receptor. Moreover, when AChR-Fc, effector cells (natural killer cells, NK92), and interleukin-2 were added to the wells with TIB-175, AChR-Fc showed dose-dependent cytotoxic activity against TIB-175. Cytotoxic activity was not confirmed when adding etanercept[a fusion protein of the tumor necrosis factor-α (TNF-α) receptor and the IgG Fc region, which has a similar structure as AChR-Fc], which suggested that the AChR α1 portion of AChR-Fc binds to the B cell receptor on AChR antibody-producing pathogenic B cells. The effector cells subsequently bind to the Fc portion of AChR-Fc, causing cellular cytotoxicity and affecting only specific pathogenic AChR antibody-producing hybridomas. Next, the binding and cytotoxic activity of AChR-Fc to patient-derived autoantibody-producing B cells were evaluated using an enzyme-linked immunospot (ELISpot) assay with peripheral blood mononuclear cells (PBMCs) from MG patients. In the absence of antigen stimulation, only some of the PBMCs from MG patients displayed spots of pathogenic B cells. However, when stimulated with pathogenic antigen AChR, the spots were increased and observed in all MG patients[14]. When AChR-Fc was administered, the spots were not detected in most MG patients. Although AChR-Fc may have an antigen stimulatory effect through its AChR region, the spots were significantly reduced following the addition of AChR-Fc, suggesting that AChR-Fc strongly damages AChR antibody-producing B cells in the PBMCs of MG patients. These results indicate that the fusion protein AChR-Fc binds to pathogenic B cells through the B cell receptor and exhibits cytotoxic activity against AChR antibody-producing B cells via the effector cells.
Experimental autoimmune MG (EAMG) is an MG animal model. In the passive transfer model of EAMG, MG symptoms are induced by administering AChR antibodies. Using this model, which was induced by the intraperitoneal administration of 1.5 mg/kg of mAb35 to 11-week-old female Lewis rats, we verified the autoantibody neutralizing activity of AChR-Fc. The EAMG rats were divided into five treatment groups: intravenous AChR-Fc (0.4, 2, or 10 mg/kg), intravenous immunoglobulin (IVIg) (800 mg/kg), or intravenous phosphate-buffered saline (PBS) at 4, 12, 24, and 32 hours following mAb35 administration. Clinical scores were evaluated from 0 to 120 hours[14]. In the control (PBS) group, muscle weakness appeared 24 hours after mAb35 administration, and the clinical score peaked at 56 hours. In contrast, in the AChR-Fc treatment group, the severity score was suppressed in a concentration-dependent manner (Fig. 2). The AChR-Fc 10 mg/kg group showed delayed symptom onset and an ameliorated severity score compared with the control group 32 hours after mAb35 administration. The therapeutic effect of AChR-Fc (10 mg/kg) was greater compared with that of IVIg treatment. AChR-Fc suppressed myasthenic symptoms in a dose-dependent manner in the passive EAMG rats, likely because of the neutralizing effect of the AChR antibodies.
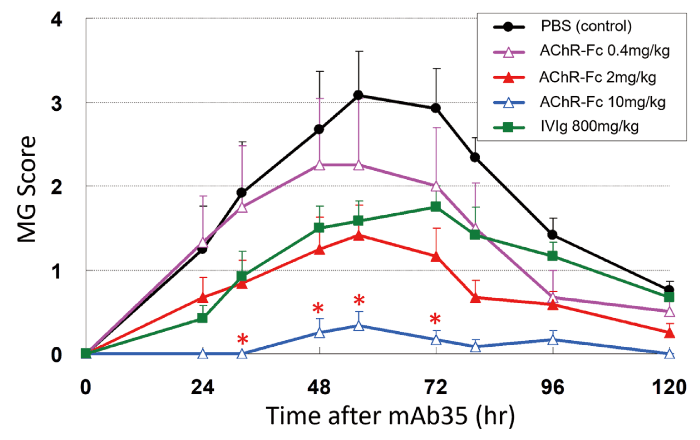
Fig. 2 Therapeutic effect of AChR-Fc in the passive EAMG model. The effect of AChR-Fc on the severity score in the passive experimental autoimmune MG (EAMG) model is shown. Modified from reference 14 (Neurotherapeutics. 2017; 14: 191-8). IVIg = intravenous immunoglobulin, PBS = phosphate buffered saline.
The active model of EAMG was induced by immunization with Torpedo AChR purified from the electroplax tissue of Torpedo californica using affinity chromatography. Compared with the passive model, the active EAMG model is similar to the pathogenesis of human MG patients because AChR antibody-producing B cells and AChR antibodies are produced from the immunized rat itself. Immunization of 8-week-old female Lewis rats with 50 μg of Torpedo AChR resulted in transient symptoms because of the excessive immune response that appeared one week following immunization. Then, the severity score gradually worsened with muscle weakness after approximately four weeks. AChR-Fc (5, 10, or 20 mg/kg) was intravenously administered to EAMG for five consecutive days from days 7, 21, and 35 after AChR immunization. In the AChR-Fc groups, a dose-dependent suppression of the severity score and a delayed onset of symptoms were observed (Fig. 3). The treatment effect was significant in the AChR-Fc 10 mg/kg and 20 mg/kg groups compared with the control group from 4.5 weeks after immunization. In addition, serum AChR antibody levels at 8 weeks were significantly lower in the AChR-Fc groups compared with those in the control group[14]. The results indicate that the AChR-Fc fusion protein exerts a dose-dependent pharmacological effect in active EAMG animal models, indicating the possibility of its clinical application. Based on the therapeutic effect in the passive and active EAMG models, the AChR antibody neutralizing effect of AChR-Fc and cellular cytotoxicity against AChR antibody-producing B cells are expected to occur in vivo[14].
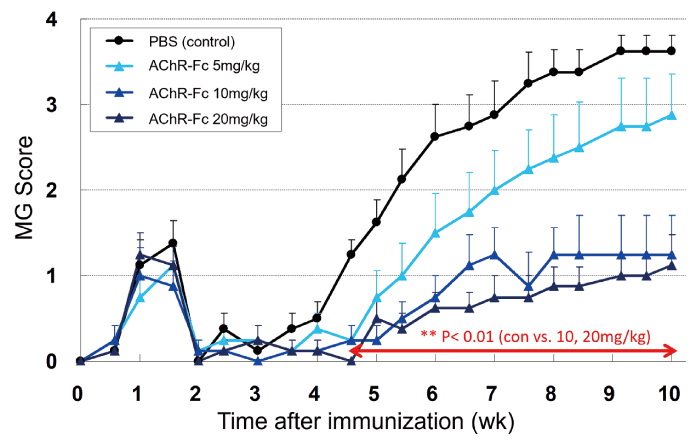
Fig. 3 Therapeutic effect of AChR-Fc in the active EAMG model. The effect of AChR-Fc on the severity score in the active experimental autoimmune MG (EAMG) model is shown. Modified from reference 14 (Neurotherapeutics. 2017; 14: 191-8). PBS = phosphate buffered saline.
The fusion protein AChR-Fc has dual mechanisms of action: neutralization of AChR antibody and cytotoxicity against AChR antibody-producing B cells. Overall, it is a promising new therapeutic candidate that may overcome AChR antibody-positive MG. Through these mechanisms, AChR-Fc is expected to have rapid, long-term, and sustained therapeutic effects. In addition, unlike steroids and immunosuppressants that suppress entire immune functions, AChR-Fc only acts on pathogenic autoantibodies and B cells, which is expected to be a safer treatment; however, there are still some unknowns about how AChR-Fc exerts its effect in vivo, which should be resolved in future studies to apply AChR-Fc in clinical practice. Regular AChR-Fc administration will be required in clinical practice, but AChR-Fc attacks and eliminates pathogenic AChR antibody-producing B cells. Therefore, its effects are expected to be longer-lasting compared with those of conventional treatments. Importantly, this Fc fusion technology may be applied to other antibody-mediated autoimmune diseases for which autoantigens have been identified and are considered to have potential for further development.
None.
None.
Not applicable.
Not applicable.
I would also like to express my sincere gratitude to Professor Satoshi Kuwabara for critically reading this manuscript.
Address correspondence to Dr. Akiyuki Uzawa.
Department of Neurology, Graduate School of Medicine,
Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba 260-8670, Japan.
Phone: +81-43-226-2129.
Fax: +81-43-226-2160.
E-mail: auzawa@chiba-u.jp
